Une nouvelle méthode quantique pour décrire les systèmes moléculaires complexes
Les calculs de structures et d'énergies sont un véritable défi quand il s'agit de tenir compte de l'environnement d'un atome ou d'une molécule. Dans ce travail, l'efficacité d'une nouvelle approche traitant séparément l'environnement et combinant deux méthodes de calculs quantiques est démontré.
La compréhension des processus physico-chimiques tels que les collisions, l'excitation par des photons ou les réactions chimiques, passe par la connaissance des énergies de liaison des électrons au sein de des atomes et nécessitent le plus souvent leur modélisation. Atomes et molécules ne sont généralement pas isolés. Par exemple, issus des embruns marins, ce sont des atomes halogènes (tels que le chlore et l'iode) contenus dans des gouttelettes d'eau qui se retrouvent impliqués dans les cycles décrivant la réactivité de la chimie de l'atmosphère. Pour décrire ces systèmes, il est nécessaire de prendre en compte un grand nombre de molécules d'eau pour simuler une gouttelette et les calculs deviennent rapidement extrêmement coûteux en temps. Dans ce travail, des chercheurs du laboratoire Physique des lasers, atomes et molécules (PhLAM, CNRS/Université de Lille) de Lille ont proposé une nouvelle approche du calcul, dite à sous-systèmes, des énergies de liaison des électrons, basée sur l'application de méthodes distinctes pour l'atome d'halogène et pour l'environnement aqueux. Ils ont ainsi obtenu une précision analogue à celles des méthodes usuelles pour la simulation des propriétés d’espèces en milieu aqueux, les méthodes dites périodiques, pour un coût nettement inférieur. Une telle approche permet en particulier de prédire le comportement des atomes les plus lourds, radioactifs ou instables, pour lesquels il est difficile de faire des expériences.
Un des systèmes considérés dans ce travail est I(H2O)50, où un atome d'iode (I) est entouré de 50 molécules d'eau (cf. figure). Il est bien connu que les atomes halogènes attirent facilement un électron pour former un anion et ce système correspond en fait à une excitation du système plus stable I-(H2O)n qui a conduit à l'éjection d'un électron. La méthode consiste tout d'abord à inventorier toutes les configurations possibles, c'est-à-dire toutes les géométries possibles des molécules d'eau entourant l'iode, à l'aide de calculs de dynamique moléculaire classique. Une centaine de configurations est ainsi sélectionnée, sur lesquelles le calcul des énergies de liaison des électrons est ensuite effectué. Ici, en raison du grand nombre de molécules d'eau nécessaire pour décrire le système, calculer les énergies de liaison des électrons avec des approches quantiques où toutes les espèces sont traitées de façon très précise, comme celle appelée EOM-IP-CCSD (Equation Of Motion-Ionization Potential-Coupled Cluster with Single and Double Excitations), conduirait à des temps de calcul prohibitifs. Les chercheurs séparent donc l'étude en deux sous-systèmes : d'une part l'atome d'iode dont les états excités sont décrits avec beaucoup de précision par la méthode EOM-IP-CCSD, et d'autre part les molécules d'eau, qui sont décrites très efficacement avec une méthode quantique appelée DFT (Density Functional Theory), adaptée pour tenir compte des propriétés spécifiques de liaison des molécules d'eau entre elles. De plus, cette méthode inclut les effets relativistes très facilement, ceux-ci étant d'autant plus importants que l'halogène est lourd.
L'approche utilisée ici est généralisable à d'autres systèmes et la bonne précision obtenue montre l’intérêt des approches quantiques à sous-systèmes pour modéliser la matière, en particulier lorsque les effets relativistes sont importants. Dans ce travail ont été étudiés tous les halogènes jusqu’à l’astate (At), un élément radioactif dont l’isotope 211 semble prometteur dans les applications de médecine nucléaire, mais qui est très difficile à étudier expérimentalement en raison de sa très courte demi-vie (7.2 heures) et pour lequel le caractère prédictif de la modélisation est donc d'un très grand intérêt.
Une nouvelle méthode quantique pour décrire les systèmes moléculaires complexes.
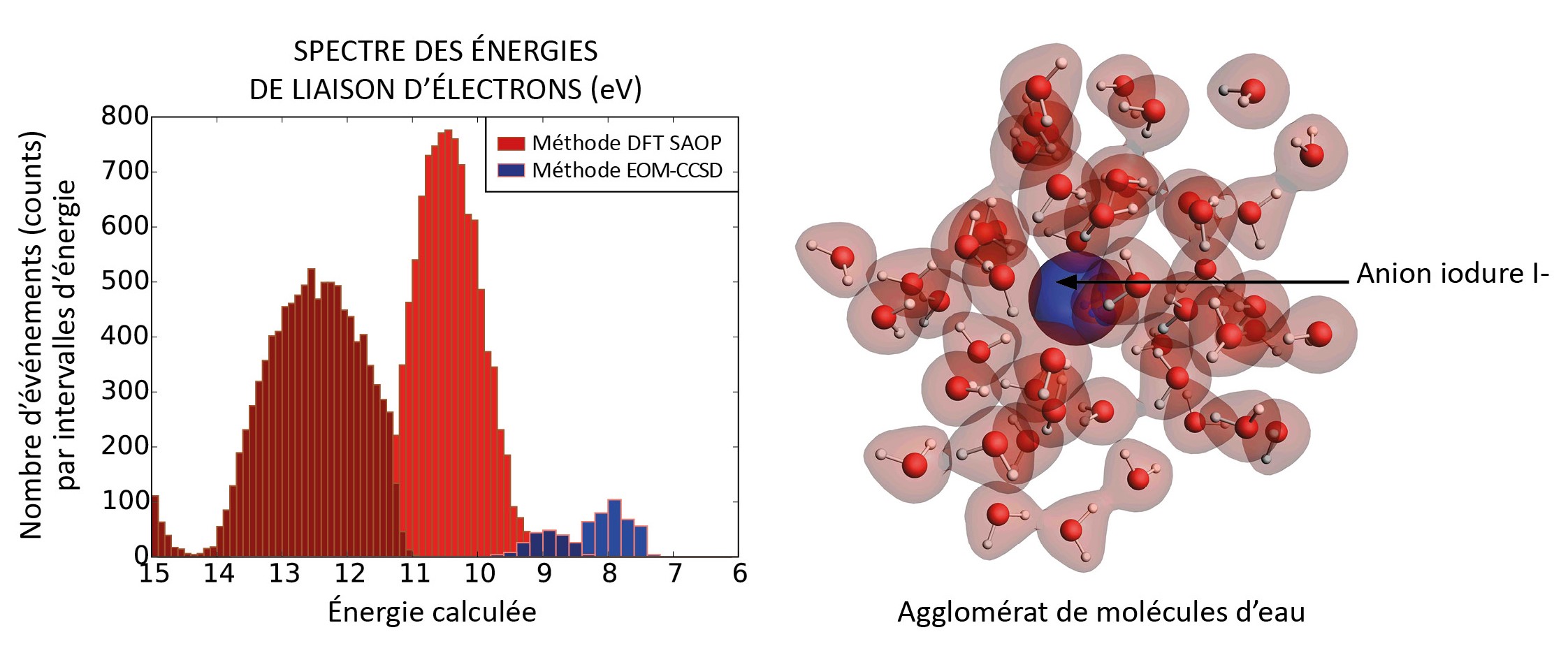
A gauche : spectre des énergies de liaison des électrons obtenue par l’approche à deux sous-systèmes.
En bleu, les énergies calculées par la méthode EOM-IP-CCSD pour l’iodure : les deux bandes autour de 8 et 9 eV correspondent au dédoublement des niveaux d'énergie dû au couplage spin-orbite.
En rouge, les énergies calculées par la méthode DFT SAOP sans couplage spin-orbite pour les molécules d’eau : les deux bandes correspondent aux états de valence d'une part (autour de 10.5 eV) et aux états plus internes d'autre part (autour de 12.5 eV). Ces calculs prennent en compte 100 configurations issues de calculs de dynamique moléculaire classique. © PhLAM (CNRS/Université de Lille)
Référence
Predictive simulations of ionization energies of solvated halide ions with relativistic embedded equation of motion coupled cluster theory. Y. Bouchafra, A. Shee, F. Réal, V. Vallet et A. Severo Pereira Gomes, Physical Review Letters, le 28 décembre 2018.
Lire l’article sur la base d’archives ouvertes ArXiv.